Processing Paired End Reads
Ok, I'm taking a break from journals. I didn't like the overly negative tone I was taking in those reviews, so I'm rethinking how I write about articles. Admittedly, I think criticism is in order for papers , but I can definitely focus on papers that are worth reviewing. Unfortunately, I'm rarely a fan of papers discussing bioinformatics applications, as I always feel like there's a hidden agenda behind them. Whether it's simply proving their application is the best, or just getting it out first, computer application papers are rarely impartial.
Anyhow, I have three issues to cover today:
The first one is pretty obvious. FindPeaks is now available under the GPL on sourceforge, and I hope people will participate in using and improving the software. We're aiming for our first tagged release on friday, with frequent tags thereafter. Since I'm no longer the only developer on this project, it should continue moving forward quickly, even while I'm busy studying for my comps.
The second point is this silly notion that keeps coming up. "How many reads were found under each peak?" I'm quite sick of that question, because it really makes no sense. Unfortunately, this was a metric produced in Robertson et al.'s STAT1 data set, and I think other people have included it or copied it. Unfortunately it's rubbish.
The reason it worked in STAT1 was because they used a fixed length (or XSET) value on their data set. This allowed them to determine the exact length of each read, which allowed them to figure out how many reads were "contiguously linked in each peak." Readers who are paying attention will also realize what the second problem is... They didn't use subpeaks either. Once you start identifying subpeaks, you can no longer assign to which peak a read spanning peaks belongs. Beyond that, what do you do with reads in a long tail? Are they part of the peak or not?
Anyhow, the best measure for a peak, at the moment at least, is the height of the peak. This can also include weighted reads, so that reads which are unlikely to contribute to a peak actually contribute less, bringing in a scaled value. After all, unless you have paired end tags, you really don't know how long the original DNA fragment was, which means you don't know where it ended.
That also makes a nice segue to my last point. There are several ways of processing paired end tags. When it comes to peak calling it's pretty simple: you use the default method - you know where the two ends are, and they span the full read. For other applications, however, there are complexities.
If the data source is a transcriptome, your read didn't cover the intron, so you need to process the transcript to include breaks, when mapping it back to the genome. That's really a pain, but it is clearly the best way to visualize transcriptome PETs.
If the data source is unclear, or you don't know where the introns are (which is a distinct possibility), then you have to be more conservative, and not assume the extension of each tag. Thus, you end up with a "tag and bridge" format. I've included an illustration to make it clear.
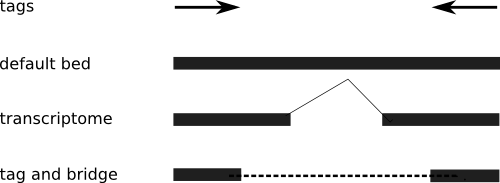
So why bring it up? Because I've had several requests for the tag-and-bridge format, and my code only works on the default mode. Time to make a few adjustments.
Anyhow, I have three issues to cover today:
- FindPeaks is now on sourceforge
- Put the "number of reads under a peak" to rest. permanently, I hope.
- Bed files for different data sources.
The first one is pretty obvious. FindPeaks is now available under the GPL on sourceforge, and I hope people will participate in using and improving the software. We're aiming for our first tagged release on friday, with frequent tags thereafter. Since I'm no longer the only developer on this project, it should continue moving forward quickly, even while I'm busy studying for my comps.
The second point is this silly notion that keeps coming up. "How many reads were found under each peak?" I'm quite sick of that question, because it really makes no sense. Unfortunately, this was a metric produced in Robertson et al.'s STAT1 data set, and I think other people have included it or copied it. Unfortunately it's rubbish.
The reason it worked in STAT1 was because they used a fixed length (or XSET) value on their data set. This allowed them to determine the exact length of each read, which allowed them to figure out how many reads were "contiguously linked in each peak." Readers who are paying attention will also realize what the second problem is... They didn't use subpeaks either. Once you start identifying subpeaks, you can no longer assign to which peak a read spanning peaks belongs. Beyond that, what do you do with reads in a long tail? Are they part of the peak or not?
Anyhow, the best measure for a peak, at the moment at least, is the height of the peak. This can also include weighted reads, so that reads which are unlikely to contribute to a peak actually contribute less, bringing in a scaled value. After all, unless you have paired end tags, you really don't know how long the original DNA fragment was, which means you don't know where it ended.
That also makes a nice segue to my last point. There are several ways of processing paired end tags. When it comes to peak calling it's pretty simple: you use the default method - you know where the two ends are, and they span the full read. For other applications, however, there are complexities.
If the data source is a transcriptome, your read didn't cover the intron, so you need to process the transcript to include breaks, when mapping it back to the genome. That's really a pain, but it is clearly the best way to visualize transcriptome PETs.
If the data source is unclear, or you don't know where the introns are (which is a distinct possibility), then you have to be more conservative, and not assume the extension of each tag. Thus, you end up with a "tag and bridge" format. I've included an illustration to make it clear.
So why bring it up? Because I've had several requests for the tag-and-bridge format, and my code only works on the default mode. Time to make a few adjustments.
Labels: FindPeaks, Open Source, paired end tags, peaks


2 Comments:
Hi Anthony,
when will your paired-end chip-seq data be publicly available?
John
Hi John,
The data itself isn't available, but all of the methods for processing it are. If that's too vague, let me know.
Anthony
Post a Comment
<< Home