ChIP-Seq normalization.
I've spent a lot of time working on ChIP-Seq controls recently, and wanted to raise an interesting point that I haven't seen addressed much: How to normalize well. (I don't claim to have read ALL of the chip-seq literature, and someone may have already beaten me to the punch... but I'm not aware of anything published on this yet.)
The question of normalization occurs as soon as you raise the issue of controls or comparing any two samples. You have to take it in to account when doing any type of comparision, really, so it's somewhat important as the backbone to any good second-gen work.
The most common thing I've heard to date is to simply normalize by the number of tags in each data set. As far as I'm concerned, that really will only work when your data sets come from the same library, or two highly correlated samples - when nearly all of your tags come from the same locations.
However, this method fails as soon as you move into doing a null control.
Imagine you have two samples, one is your null control, with the "background" sequences in it. When you seqeunce, you get ~6M tags, all of which represent noise. The other is ChIP-Seq, so some background plus an enriched signal. When you sequence, hopefully you sequence 90% of your signal, and 10% of the background to get ~8M tags - of which ~.8M are noise. When you do a compare, the number of tags isn't quite doing justice to the relationship between the two samples.
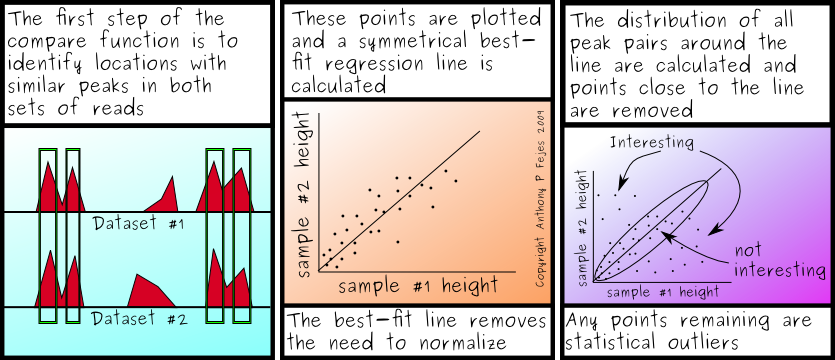
So what's the real answer? Actually, I'm not sure - but I've come up with two different methods of doing controls in FindPeaks: One where you normalize by identifying a (symmetrical) linear regression through points that are found in both samples, the other by identifying the points that appear in both samples and summing up their peak heights. Oddly enough, they both work well, but in different scenarios. And clearly, both appear (so far) to work better than just assuming the number of tags is a good normalization ratio.
More interesting, yet, is that the normalization seems to change dramatically between chromosomes (as does the number of mapping reads), which leads you to ask why that might be. Unfortunately, I'm really not sure why it is. Why should one chromosome be over-represented in an "input dna" control?
Either way, I don't think any of us are getting to the bottom of the rabbit hole of doing comparisons or good controls yet. On the bright side, however, we've come a LONG way from just assuming peak heights should fall into a nice Poisson distribution!
The question of normalization occurs as soon as you raise the issue of controls or comparing any two samples. You have to take it in to account when doing any type of comparision, really, so it's somewhat important as the backbone to any good second-gen work.
The most common thing I've heard to date is to simply normalize by the number of tags in each data set. As far as I'm concerned, that really will only work when your data sets come from the same library, or two highly correlated samples - when nearly all of your tags come from the same locations.
However, this method fails as soon as you move into doing a null control.
Imagine you have two samples, one is your null control, with the "background" sequences in it. When you seqeunce, you get ~6M tags, all of which represent noise. The other is ChIP-Seq, so some background plus an enriched signal. When you sequence, hopefully you sequence 90% of your signal, and 10% of the background to get ~8M tags - of which ~.8M are noise. When you do a compare, the number of tags isn't quite doing justice to the relationship between the two samples.
So what's the real answer? Actually, I'm not sure - but I've come up with two different methods of doing controls in FindPeaks: One where you normalize by identifying a (symmetrical) linear regression through points that are found in both samples, the other by identifying the points that appear in both samples and summing up their peak heights. Oddly enough, they both work well, but in different scenarios. And clearly, both appear (so far) to work better than just assuming the number of tags is a good normalization ratio.
More interesting, yet, is that the normalization seems to change dramatically between chromosomes (as does the number of mapping reads), which leads you to ask why that might be. Unfortunately, I'm really not sure why it is. Why should one chromosome be over-represented in an "input dna" control?
Either way, I don't think any of us are getting to the bottom of the rabbit hole of doing comparisons or good controls yet. On the bright side, however, we've come a LONG way from just assuming peak heights should fall into a nice Poisson distribution!
Labels: Bioinformatics, Chip-Seq, false discovery rate, FindPeaks, Vancouver Short Read Analysis Package